


4月15日,認監委官網發布口罩等防疫用品出口歐盟準入信息指南(第二版)。

一、CE標志制度
CE標志制度是歐盟對產品進入歐盟市場進行的監管方式。加貼CE標志的產品表明產品符合歐盟有關安全、健康、環保等法規要求,可以在歐盟27個成員國、歐洲貿易自由區的4個國家、以及英國和土耳其合法上市銷售。按照歐盟規定,不同產品采用不同的評價方式加貼CE標志,主要有兩種方式:絕大部分產品是制造商采取自我符合性聲明方式,就可以加貼CE標志;部分風險相對更高的產品需要經過歐盟授權的第三方機構,即公告機構(Notified Body)進行符合性評定后,方可加貼CE標志。產品經公告機構符合性評定后加貼CE標志的流程:產品制造商向公告機構提出申請,公告機構為制造商提供符合性評定服務,制造商及產品符合法規要求的,向制造商發放CE證書。制造商依據CE證書簽署符合性聲明,產品加貼CE標志后就可以進入歐盟市場。下表列舉了歐盟、成員國的主管當局、公告機構和制造商的職責。
|
角色 |
職責 |
|
歐盟 (EU) |
制定歐盟法規(指令)、協調各成員國的活動。 |
|
成員國主管當局 |
負責對應法規(指令)在本國的執行、聯合歐盟對公告機構進行授權,監督公告機構行為,并進行產品的上市后監督。 |
|
公告機構 (NB) |
進行符合性審核,發放CE證書。 |
|
制造商 Manufacturer |
確保自己的產品滿足歐盟法規(指令)的要求,進行自我聲明或者申請公告機構符合性評定,加貼CE標志,并對產品質量安全負責。 |
口罩在歐盟根據預期用途的不同,分為醫用口罩和個人防護口罩兩種,分別歸屬醫療器械條例EU2017/745(MDR)或醫療器械指令93/42/EEC(MDD)和個人防護設備條例EU2016/425(PPE)進行管理。如何判定具體產品屬于哪一種口罩,需參照對應的法規規定和標準要求。
|
中國 |
歐盟 |
|
日常防護性口罩 GB/T32610-2016 |
個人防護口罩EN 149:2001+A1:2009 Respiratory protective devices; 分為FFP1, FFP2 and FFP3三類 |
|
呼吸防護口罩GB 2626-2006;過濾效率分為三級KN90 (90%),KN95 (95%),KN100(99.97%) |
|
|
醫用防護口罩GB19083-2010;過濾效率1, 2, 3級:95%,99%,99.97% |
醫用口罩 EN 14683:2019 Medical face masks;分為Type I (95%),Type II (98%),Type IIR (98%). * (EN 14683:2005 名稱是手術口罩Surgical masks) |
|
醫用外科口罩YY 0469-2011 |
|
|
一次性使用醫用口罩 YY/T0969-2013 |
1.技術文件要求:
參照MDR法規附錄II和附錄III的要求(MDD為附錄7),技術文件通常包括以下七個部分: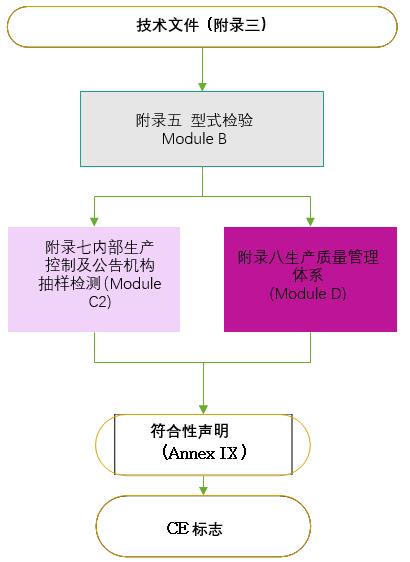
|
口罩類別 |
醫用非滅菌口罩 |
醫用滅菌口罩 |
個人防護口罩 |
|
用途 |
在醫療環境中佩戴以保護病人,或者由病人佩戴,防止疾病傳播 |
在手術環境或類似的醫療環境中佩戴以保護病人或者環境,防止疾病傳播 |
個人佩戴以保護該人員的健康和安全,免除對應的風險 |
|
適用法規 |
MDR法規或MDD指令 |
MDR法規或MDD指令 |
PPE法規 |
|
分類 |
I類非滅菌 |
I類滅菌- Class Is |
III類 |
|
主要適用標準 |
EN 14683 |
EN 14683 + 滅菌標準 |
EN 149 |
|
技術文件 |
需要 |
需要 |
需要 |
|
體系要求 |
售后監督要求 |
需要 |
需要 |
|
符合性評定途徑 |
自我聲明 |
公告機構評定 |
公告機構評定 |
|
CE證書 |
不需要 |
一張 |
兩張 |
|
符合性聲明 |
需要(在確保產品安全有效符合法規要求后簽署) |
需要(在獲取CE證書后簽署) |
需要(在獲取CE證書后簽署) |
|
CE標志 |
需要 |
需要(CE +四位數的公告機構號碼) |
需要(CE +四位數的公告機構號碼) |
 微信公眾號
微信公眾號 微信咨詢
微信咨詢



 信息提交成功
信息提交成功